A dive into blood-brain barrier permeability prediction models (part 3)
- Anastasiia Navalikhina
- Oct 12, 2023
- 5 min read

In the previous articles, we described the problem of Brain Blood Barrier (BBB) permeability and the importance of considering this feature in the drug development process. We also presented the need for constructing ML models that predict substrates and inhibitors of two known BBB influx transporters, OATP1A2 and OATP2B1.
In this article, we describe how we constructed six models that classify drugs into :
substrates/non-substrates of BBB transporters
inhibitors/non-inhibitors of OATP1A2
inhibitors/non-inhibitors of OATP2B1.
We also briefly discuss the molecular features that determine the potential of being a substrate or inhibitor of transporters for different drugs. We define the relationship between these features and the property of being substrate or inhibitor using linear Pearson correlation and non-linear Maximum Information Coefficient (MIC).
Predicting substrates of OATP1A2 and OATP2B1
As we discussed in our previous article, the substrates class is minor, and it needs to be oversampled to restore the balance in the data set and build models which can define the substrates class. We added 102 positive class synthetic points to the data set, which shifted the ratio of classes from 0.09 to 0.5. Next, two types of models were built to classify drugs as substrates/non-substrates of BBB transporters: Random Forest Classifier and Principal Component Regressor.
The first model is Random Forest Classifier (RF) with modified class weights: the weight of the substrates class was set to be 5 times greater than that of the non-substrates class. This implies that the substrate instances have a higher cost for misclassification. As a result, we will have all true substrates and some amount of false positives in the predicted class. In the perfect case, the classifier will have a maximum substrate recall with a slightly lower precision value.
The metrics for the constructed RF Classifier were satisfying: for the training set, substrates class precision value was not critically affected when we set the thershold to obtain a 100 % recall. For the test and validation sets, we obtained 92% and 87% recall for the positive class. This means that in the train set all substrates were classified correctly, and in the test and validation sets there were no more than 13 % of molecules misclassified as non-substrates (Table 1 below).
The second model used for substrates classification is Principal Component Regression (PCR) which is PCA transformation followed by a Logistic Regression. We used eight principal components to build our model and modified class weights of the Logistic Regression so the weight of the substrates class was 5 times greater than that of non-substrates. The PCR model failed to distinguish substrates from non-substrates, as with getting high recall (~ 90 %), the precision drops below 60 % in train and test sets. This means that too many non-substrates would be classified as substrates, so this model generates too many false positives. In the validation set both precision and recall have too low values, indicating this model predictions to be nearly random.

Table 1 - RF and PCR perf. metrics for substrates
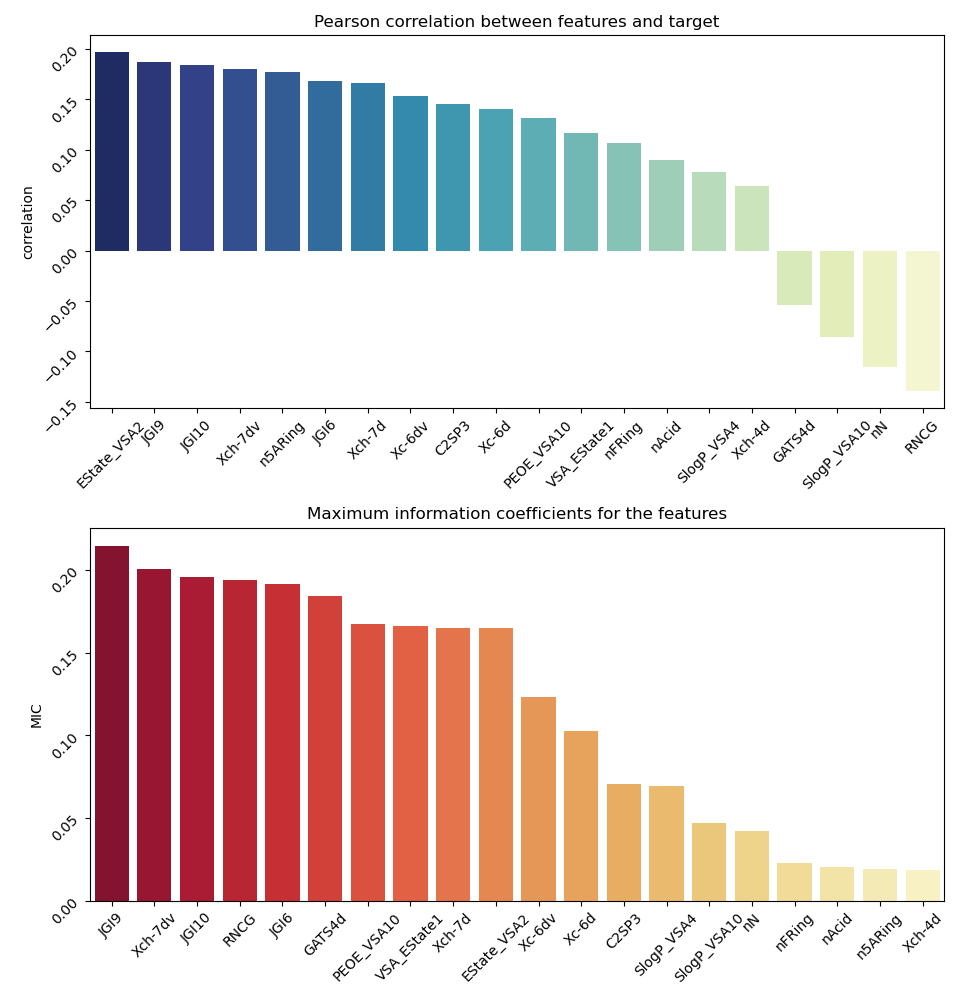
Fig. 1- Pearson and MIC correlations of the
relevant molecule descriptors for substrates
We calculated linear and non-linear relationships between the property of being a substrate and molecular features of a drug that the model uses for classification (Fig. 1). To make sense of these descriptors, we will briefly discuss some of them. First, we can see that there are three descriptors in the top-10 related to the JGI family descriptors, which are the mean topological charge of a molecule. These are quantitative descriptors of the area through which the compound is interacting with other molecules and the charge distribution on this area. There are also VSA descriptors, which are van der Waals surface area descriptors for the amount of area having certain property: PEOE_VSA - the amount of VSA with partial charges property, SlogP_VSA - the amount of VSA with water/octanol solubility coefficient property. We can assume that the topological charge of the molecule and its solubility are important features defining the ability of the molecule to pass through either OATP1A2 or OATP2B1 BBB channels.
Predicting inhibitors of OATP1A2
In order to build models for OATP1A2 inhibitors identification, first we resampled inhibitors minority class and added 12 synthetic points, so the transformed data set have a 1:1 class ratio. After that, two models were built to classify drugs into inhibitors and non-inhibitors. First was a Random Forest with a modified class weight for inhibitors, two times higher than for non-inhibitors. Similar to the substrates classification model, RF classification performed well, giving about 100 % recall for class 1 in both train and validation sets, but lower precision for class 0 compared to the substrates predicting model (Table 2).
As for substrates detection, we build the second model, Principal Component Regression, for detecting OATP1A2 inhibitors. The PCR model is constructed with eight principal components and the Logistic Regression model in PCR has a weight ratio of 1:5 (inhibitors class to non-inhibitors class). After training, it shows comparable to the RF metrics for training and test data sets but fails in all scores for the validation set.

Table 2 - RF and PCR perf. metrics for OATP1A2 inhibitors
Some of the descriptors used to build models have moderate values of Pearson’s R and MIC with the target value. Among these are MATS1se, GATC3c, and ATSC8c (Fig. 2). The families of MATS- (Moran autocorrelation), ATSC- (Moreau-Broto autocorrelation), and GATS- (Geary autocorrelation) are descriptors that show how certain property is distributed along the molecule topological structure. So, as for substrates, some properties of molecule topology are important determinants of its ability to be an OATP1A2 inhibitor.

Fig. 2- Pearson and MIC correlations of the
relevant molecule descriptors for OATP1A2 inhibitors
Predicting inhibitors of OATP2B1
As in the substrates data set and in the OATP1A2 inhibitors data set, we have a lower number of molecules known to be OATP1B2 inhibitors than those which are known to be non-inhibitors. To rebalance data, we resampled data points from inhibitors class to have class ratio 1:1 by creating 151 synthetic instances.
As in previous cases, two models were built to classify drugs into inhibitors and non-inhibitors of OATP2B1
Random Forest with modified class weights. The weights ratio is 1:5
Principal Component Regression with fifteen principal components used to build a model and weights ratio 1:5.
Evaluation of both models’ performance is shown in Table 3. As with previous models, RF succeeded in keeping high precision values with elevated recall, and PCR failed to do so.

Table 3 - RF and PCR perf. metrics for OATP2B1 inhibitors
For the features used by models to define the ability of drugs to inhibit OATP2B1, the values of Pearson’s coefficient and MIC are not higher than 0.35. Despite this, among the most important features we can see MATS-, GATS-, and ATSC- descriptors similar to that we saw for OATP2A1 inhibitors predicting models, and also VSA descriptors we saw for predicting substrates (Fig. 3). So, comparable properties of a drug define whether it can bind and suppress the activity of OATP1A2 or OATP2B1.

In the end, we see that Random Forest models are more suitable for drug classification than PCR. For all three drug activity predictions, RF succeeded to keep high values of precision for increased positive class recall.
We can also conclude, that for all the constructed models which predict, the topological structure and charge distribution of the drug defines whether it can bind to the transporter and become its’ substrate or inhibitor.
--------------------------------
* Follow us on LinkedIn for next blog updates:
* Interested in our skills? Let's discuss your projects together:

or
* Our public Github repository:

--------------------------------
References
Roberto Todeschini and Viviana Consonni Molecular Descriptors for Chemoinformatics
https://www.epa.gov/sites/production/files/2015-05/documents/moleculardescriptorsguide-v102.pdf